What Is Quality Risk Management [QRM]?

Quality Risk Management (QRM) is used in product quality assurance and as a key element for compliance-risk management is essential for an organization to maintain their quality systems.
QRM should be a vital component of an organization’s quality system and best practices to achieve higher levels of process control and to manage and control risks to product quality and patient safety.
This process helps today’s manufacturers demonstrate that their system or process is suitable for its intended use. QRM is also a business-critical process as it helps manage and control an enterprise’s business and compliance-risk.
What Is Quality Risk Management?
Quality risk management is a systematic, risk-based approach to quality management. The process is composed of the assessment, control, communication, and review of quality and compliance risks. It is especially critical in the pharmaceutical industry, where product quality can greatly affect consumer health and safety.
Current global GMP regulations require that manufacturing processes be designed and controlled to assure that in-process materials and the finished product meet predetermined quality requirements and do so consistently and reliably as demonstrated through Process Validation.
How To Create A QRM Process
A well-written QRM procedure that describes the risk management process requirements and step-by-step instructions is essential to ensure that an organization can easily, repeatedly, and consistently comply with QRM policy requirements and regulator expectations.
The QRM process flow defined by ICH Q9, Quality Risk Management, is illustrated below.
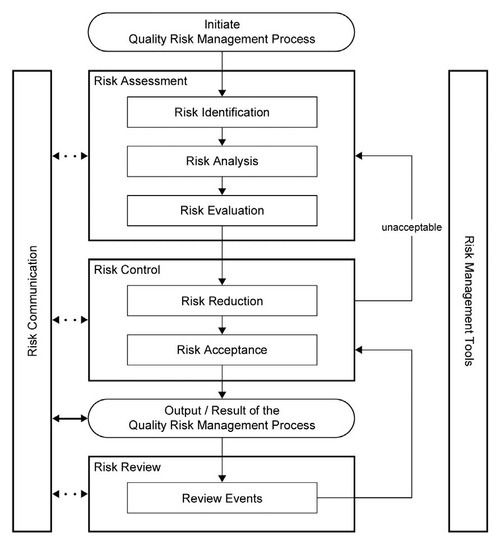
How Are Risk Management Principles Used?
Utilizing risk management principles (an equally important GEP) to evaluate potential risks of design solutions during the design, specification, and verification process will help deliver manufacturing systems that are right the first time and will help ensure that systems are suitable for their intended use.
Utilizing quality risk management practices during the design and engineering phase of a project is a regulatory expectation to manage risk associated with product quality and patient safety. Product and process information related to product quality and patient safety should form the basis of science and risk-based decisions to ensure that manufacturing systems are designed and verified as fit for the system’s intended use.
Unacceptable risks to product quality and patient safety should be reduced to an acceptable level or eliminated, as much as possible, prior to process validation. Risk management activities should be performed by subject matter experts and key stakeholders at appropriate stages of a product’s lifecycle.
How To Document Risk Assessments
Documenting risk assessments is a good engineering practice and an effective management tool for identifying, analyzing, and evaluating risks associated with a specific system or process step. The risk assessment process should identify and focus on critical aspects that may impact critical quality attributes and critical process parameters.
A risk assessment should address three fundamental questions when defining and analyzing a risk:
- What could go wrong?
- What are the consequences (severity)?
- What is the likelihood (probability) it will go wrong?
Note: This article does not cover risk analysis techniques, such as Failure Mode and Effect Analysis (FMEA), Failure Mode Effect and Criticality Analysis (FMECA), Fault Tree Analysis (FTA), Hazard and Operability Study (HAZOP), Fishbone Analysis, etc., that may be used during risk assessment activities.
Why Use A Risk Assessment?
Risk assessment activities should be followed by taking action on design improvements, implementing risk reduction and control mechanisms (i.e., automation, process and / or procedural controls, etc.) or accepting risks to the manufacturing system. Assessing and controlling risks ultimately leads to reducing process variability and improving process capability.
The level of effort, formality, and documentation (i.e., risk communication) should be commensurate with the level of risk or proportional to the complexity of the system. For example, the risk assessment for a non-complex change to an existing system may be documented in a brief summary as part of the change control process documentation.
Implementing a new complex manufacturing system or computer system may require a standalone risk assessment document. The level of formality or documentation required for a system can also be based on system impact (i.e., indirect impact systems require less formality or documentation).
Understanding URS Documents
A URS is a planning document used whenever a business is planning on acquiring a different system or even strategizing to enhance their current systems. The development of an URS should be supported by “Risk Management” activities. The critical aspects, critical quality attributes, and critical process parameters provide key ‘process knowledge and understanding’ required to perform an informed assessment of risks that will ultimately need to be mitigated and / or controlled for a manufacturing system.
As a system’s design evolves, changes to user requirements and any associated new risks should be updated in a single version-controlled URS document. This practice ensures that risks are documented and controlled and subsequently verified during qualification of the final delivered solution.
Why Implement QRM?
In 2015, the FDA published a document entitled “Pharmaceutical Quality for the 21st Century A Risk-Based Approach Progress Report.“ This FDA initiative presents a ‘risk-based’ philosophy for the agency and industry to use in establishing practices and policies for managing manufacturing science and quality into the future.
Today’s pharmaceutical and medical device manufacturers must comply with strict global regulations and meet global regulator expectations for controlling risks that could impact product quality and patient safety. Failure of pharmaceutical manufacturers to comply with global regulators’ expectations may result in a variety of negative business-related impacts including:
- Withheld distribution of product
- Lost revenue
- Falling stock prices
- Delayed product approvals
- Fines
- Product recalls
- Litigation
- Consent decrees
- Warning letters
- Adverse affects or physical harm to consumers
Industry executives and managers are challenged with creating strong, compliance-centric cultures within an ever-expanding complex global organizational and regulatory structure. Many executives and managers see the wisdom in Aristotle’s quote, ‘”We are what we repeatedly do.”‘
These executives recognize ‘Compliance’ differentiated from ‘Quality’ as a critical competency for maintaining their company’s competitive position and more importantly as a key factor for assuring the highest quality in their manufactured products.
In today’s competitive business world, if a manufacturing operation does not effectively manage risk and have a robust Quality System that is consistently adhered to, the operation will not long prosper. QRM is an essential tool in management’s ‘operational excellence and compliance-risk management toolkit’ that helps build strong, compliance-centric cultures.
QRM Related Keywords You Should Know
The following keywords are ones that you will see pop up here and there when diving into QRM.
Critical Aspects
Functions, features, abilities, and performance or characteristics necessary for the manufacturing process and systems to ensure consistent product quality.
Critical Quality Attribute
A property of a product or output of the process that is reflective of the process performing as expected. Critical quality attributes correlate to critical process parameters.
Critical Process Parameter
A process parameter that can affect the critical quality attribute (s) of the product / process / unit that must be controlled within a pre-determined range or specification criteria.
Risk
The combination of the probability of occurrence of harm and the severity of that harm.
Risk Assessment
A systematic process for organizing information to support a risk decision that is made within a risk management process. The process consists of the identification of hazards and the analysis and evaluation of risks associated with exposure to those hazards.
Risk Management
The systematic application of quality management policies, procedures, and practices to the tasks of assessing, controlling, communicating, and reviewing risk.
How Will You Approach Quality Risk Management?
QRM will help manufacturers meet regulatory expectations for validation of critical processes and systems. The FDA and industry thought-leaders believe that modern quality systems, when coupled with manufacturing process and product knowledge and the use of effective risk management practices, should allow a firm to make changes to facilities, equipment, and processes without the need for prior regulatory approval.
Manufacturers with a robust quality system and appropriate process knowledge can implement many types of improvements that will lower the risk of manufacturing problems and may lower the need for regulatory oversight which will result in shorter and fewer FDA inspections.